Representative Tumor Sampling Method Lessens Molecular Analyses Bias
By LabMedica International staff writers
Posted on 25 May 2020
Over the past decade, clinical scientists have demonstrated the inability of an individual biopsy or formalin-fixed paraffin-embedded (FFPE) block to capture the genetic diversity of a solid tumor. Posted on 25 May 2020
When pathologists extract tumor tissue to identify potential biomarkers for downstream therapy, they deal with issues related to tumor heterogeneity. By analyzing a single sample from a fixed location, they do not always get a representative sample of the cancer, and a tissue biopsy usually only presents less than 0.0005% of the tumor.
.")
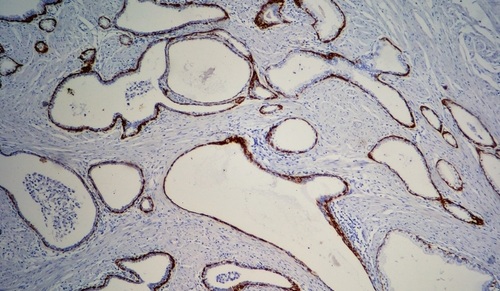
Image: Schematic diagram of how the representative tumor sampling method lessens bias in molecular analyses (Photo courtesy of Francis Crick Institute).
Clinical Scientists at the Francis Crick Institute (London, UK) and at Roche Tissue Diagnostics (Tucson, AZ, USA) and their international colleagues have developed a method that they believe could improve clinical tumor molecular profiling by generating accurate tumor mutation burden scores. The team first analyzed data for 1,667 samples across six tumor types from the National Cancer Institute's Cancer Genome Atlas (TCGA). For each sample, they assessed how much of each tumor was sampled.
The system, called Rep-Seq requires at least 1 gram of patient leftover tissue following pathology extraction. The process starts with a pathologist macro-dissecting a sample to remove normal tissue that is at least 5 cm away from the tumor. Tumor and normal tissue are separately incubated and then blended with a specific solution inside a homogenizer. Tumor and immune cells are further separated using fluorescence-activated cell sorting, enriching the representative sample for tumor cells. The group found that tumor sequencing protocols had a high under-sampling bias. Sampling bias was likely also affected by the levels of heterogeneity and purity of tumor cells in the sample.
To determine the effect of spatial bias in single-biopsy sampling, the team pooled extracted DNA from 1,184 multi-region biopsies, taken from 79 primary renal carcinomas (RCCs), to create "cocktail samples." Subjecting the samples to next-generation sequencing (NGS), the team compared mutation calls with previously generated single-biopsy and multi-region biopsy data. They found that the cocktail samples discovered all of the true-set mutations, compared with single biopsies, which only discovered 73%. The team therefore believes that a more representative sample can lead to improved variant detection.
The investigators used tumor masses from leftover material post-surgery, and then applied Rep-Seq on 11 tumors from breast, lung, colorectal, and RCC cancers. Choosing a large clear cell RCC tumor (RS1), the team collected 68 fresh-frozen biopsies from the primary tumor and later homogenized the rest using Rep-Seq. The team performed whole-exome sequencing (WES) on seven biopsies and the Rep-Seq sample, identifying 76 unique mutations. They then compared the Rep-Seq results to the single-biopsy regions and found that the variant allele frequencies (VAFs) from Rep-Seq closely matched the overall tumor VAFs in the 68 biopsies. The group only failed to detect three of the 76 mutations in the Rep-Seq sample.
Samra Turajlic, MBBS, PhD, a Consultant Medical Oncologist and corresponding author of the study, said, “Taking a small sample from a solid cancer, which contains millions or billions of cells, is very problematic, as you deal with the issue of reproducibility. Beyond seeing if there's enough leftover tumor to create a homogenized sample, we wanted to see if the sample will allow you to predict both prognostic and therapeutic markers robustly and accurately.” The study was published on May 5, 2020 in the journal Cell Reports.

Gold Member
Nucleic Acid Extractor System
NEOS-96 XT


Automated Clinical Chemistry Analyzer
Envoy 500+

Chromogenic Culture System
InTray™ COLOREX™ ECC