Lab-Developed Tests Compared with Approved Diagnostics
By LabMedica International staff writers
Posted on 30 Jan 2018
The debate about the role of the government-approved diagnostics in the regulation of laboratory-developed tests (LDTs) has focused attention on the analytical performance of all clinical laboratory testing.Posted on 30 Jan 2018
This is particularly important when it comes to detecting common types of cancer mutations such as the B-Raf proto-oncogene, serine/threonine kinase (BRAF), the epidermal growth factor receptor (EGFR), and Kirsten rat sarcoma virus (KRAS) oncology analytes.
.")
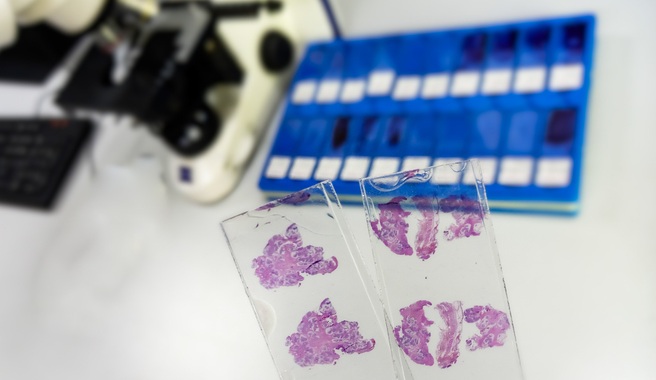
Image: The FDA-approved BRACAnalysis CDx test as a companion diagnostic (Photo courtesy of Myriad Genetics).
A team of scientists from different institutions and led by those at (Brigham and Women's Hospital, Boston, MA, USA) compared the performance of LDTs and US Food and Drug Administration (FDA, Silver Springs, MD, USA) approved companion diagnostics (FDA-CDs), in proficiency testing (PT) provided by the College of American Pathologists Molecular Oncology Committee (CAP, Northfield, IL, USA). The team analyzed the performance of 6,897 tests from laboratories participating in the CAP Proficiency Testing (PT) for BRAF, EGFR, and KRAS, oncology analytes used often by both LDTs and FDA-CDxs. A total of 6,897 PT responses were included: BRAF (n = 2,524; 14 PT samples), EGFR (n = 2,216; 11 PT samples), and KRAS (n = 2,157, 10 PT samples). FDA ompanion diagnostics and LDTs are compared for both accuracy and preanalytic practices of the laboratories.
Using CAP standards, they compared accuracy and preanalytic practices of the laboratories and found that both types of tests exceeded 97% accuracy across the three cancer genes. In another important finding, the scientists discovered that at least 60% of the participating laboratories had adapted an FDA-CDx test to the point that it changed the classification to an LDT. According to the specialists, laboratories did this to “allow for a greater breadth of sample types, minimum tumor content, and instrumentation.” From a regulatory perspective, the data suggest that there’s not as much of a difference between FDA-CDxs and LDTs as previously thought. The high marks on LDT proficiency are significant, given the scrutiny these tests have experienced over their reliability. FDA for some time has advocated for increased oversight of LDTs, claiming that they should be regulated as medical devices.
Annette S. Kim, MD, PhD, an associate pathologist and first author of the study, said, “These modifications appear to be driven by the exigencies of real day-to-day clinical practice that requires altering the assays to meet the needs of a variety of clinical situations that may not be accommodated by the FDA-approved protocol.” The study was published originally published online on December 14, 2017, in the journal JAMA Oncology.
Related Links:
Brigham and Women's Hospital
US Food and Drug Administration
College of American Pathologists

Gold Member
Quantitative POC Immunoassay Analyzer
EASY READER+

Electrolyte Analyzer
BKE-B

Steam Sterilizer
Hi Vac II Line